Clair C. Patterson Award, 2016

It is our great honor to announce that Dr. William H. Casey has won the Clair C. Patterson Award for 2016 from the Geochemical Society, for his contributions to geochemical science, most notably his work on high-pressure solution NMR! As a recipient, Dr. Casey travelled to Yokohama, Japan for Goldschmidt 2016! Dr. Casey is pictured here with his son, James, after accepting the medal.
The award is given in honor of Dr. Clair C. Patterson, who developed the uranium-lead dating system that was used to calculate the age of the Earth at 4.55 billion years. More can be found out about the award (as well as previous recipients) here.
Welcome to our CSMC Summer Interns!

The Casey lab welcomes all of our CSMC SmartCamp interns!! They are here as part of the Center for Sustainable Materials Chemistry (CSMC) in an effort to foster collaboration and to gain experience in a laboratory setting. Please welcome Jesus M. Lopez Baltazar, Derek Popple, and Nichole Rogovoy and welcome back Lily Gordon!
Happy Holidays from the Casey Lab!

Happy Holidays from the Casey lab!!
Mary Collins visits the lab!

Mary Collins joined as an honorary member in 2015. She is currently a Post-Doctoral Scholar at LBL.
Lily Gordon joins the lab!

Lily Gordon joined Casey lab in 2015 as an undergraduate researcher.
Joseph Callahan joins the lab!

Joseph Callahan joined Casey lab in 2015 as an undergraduate researcher.
Noushin Akhavantabib joins the lab!

Noushin Akhavantabib joined Casey lab in 2015 as an undergraduate researcher.
Anna Oliveri joins the lab!

Anna Oliveri joined Casey lab in 2015 as a post-doctoral scholar.
Elaine Villena joins the lab!

Elaine Villena joined Casey lab in 2014 as a graduate student.
Gerry Ochoa joins the lab!

Gerry Ochoa joined Casey lab in 2014 as a graduate student.
Brent and Jeff attend ENC 2014 in Boston

Brent presented his work on the development of the high-pressure NMR probe in the Exotica Session of the conference. Here Brent and Jeff are about to enjoy a cruise of the Boston Harbor, sponsored by Bruker BioSpin.
Corey Pilgrim joins the lab!

Corey Pilgrim joined Casey lab in 2014 as a graduate student.
Brent Pautler joins the lab!

Brent Pautler joined Casey lab in September 2013 as a postdoc after finishing his PhD degree from University of Toronto. He is working on high pressure NMR project.
Lizhi Tao joins the lab!

Lizhi Tao joined Casey lab in 2013 as a graduate student pursuing a Ph.D degree.
Lawrence Scholar Program
27th of November, 2012

Congratulations to Adele Panasci for being selected to participate in the Lawrence Scholar Program! Under this prestigious fellowship Adele will continue her work with 237Np at the Seaborg Institute at Lawrence Livermore National Lab.
Synthesis and characterization
of a soluble vanadium-containing Keggin polyoxoniobate via ESI-MS and 51V NMR: (TMA) 9[V3Nb12O42] ·18H2O
28th of October 2012

Abstract: The vanadium-containing heteropolyoxoniobate (TMA)9[V3Nb12O42]·18H2O was synthesized by hydrothermal reaction of V2O5 and hydrous niobium oxide in tetramethylammonium hydroxide solution. The cluster has an α- Keggin structure with a central VO4 and two trans-bicapped VO5. The water-soluble product was characterised by X-ray crystallography, ESI-MS and both liquid- and solid-state 51V NMR. The solid- and solution-phase 51V NMR spectra indicate two major peaks corresponding to the one VO4 and two VO5 sites.
Son, J.-H., Ohlin, C. A., Larson, E. C., Yu, P., Casey, W. H. Synthesis and characterization of a soluble vanadium-containing Keggin polyoxoniobate via ESI-MS and 51V NMR: (TMA) 9[V3Nb12O42]·18H2O. Eur. J. Inorg. Chem., Accepted.
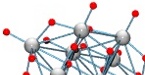
A new class of soluble and stable
transition-metal-substituted polyoxoniobate: [CrIII2(OH)4Nb10O30] 8-
4th of September, 2012
Abstract: Hydrothermal synthesis of [CrIII2(OH)4Nb10O30]8- in gram-scale quantities leads to a new polyoxometalate ion composed of two CrNb6O19 Lindqvist-type units that are fused via shared faces. The two CrIII atoms are located in the centre of the molecule and are bridged by two µ4-O atoms. Electronic transitions are calculated using density functional theory and compare well with the measured UV-Vis spectra.
Son, J.-H., Ohlin, C. A., Casey, W. H. A new class of soluble and stable transition-metal-substituted polyoxoniobate: [Cr2(OH)4Nb10O30]8-. Dalton Trans., 2012, 41, 12674-12677.
Welcome Will Elliott!
25th of June, 2012

Will Elliott, an undergraduate at the University of Oregon, joins the Casey Lab, as well as the Navrotsky Lab, to participate in undergraduate summer research!
Welcome Emma Larson!
25th of June, 2012

Emma Larson, an undergraduate at Saint Olaf College, joins the Casey Lab to participate in undergraduate summer research!
Outstanding Chemistry Dissertation Award
8th of June, 2012

Congratulations to Greg McAlpin for receiving the Outstanding Chemistry Dissertation Award! This prestigious awad is only given once per year. Greg presented his research talk, entitled "CW and Pulse EPR Studies of Colbalt-Catalyzed Water Oxidation," on Friday June 8, 2012, after which he received his award. His talk highlighted the energy needs of the world, focusing on solar as a primary source of energy. He presented an overview of his work and focused on characterizing colbalt catalysts that split water to store solar energy.


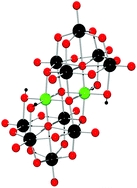
Rates of Water Exchange on the
[Fe4(OH)2(hpdta)2(H2O)4] 0 Molecule and Its Implications for Geochemistry
23th of May, 2012
Abstract: The ammonium salt of [Fe4O(OH)(hpdta)2(H2O)4]- is soluble and makes a monospecific solution of [Fe4(OH)2(hpdta)2(H2O)4]0(aq) in acidic solutions (hpdta = 2-Hydroxypropane-1,3-diamino-N,N,N',N'-tetraacetate). This tetramer is a diprotic acid with pKa1 estimated at 5.7±0.2, pKa2 = 8.8(5)±0.2. In the pH region below pKa1, the molecule is persistent in solution and 17O NMR line widths can be interpreted using the Swift-Connick equations to acquire rates of ligand substitution at the four isolated bound waters. Averaging five measurements at pH<5, where contribution from the less-reactive conjugate base are minimal, we estimate: kex298 = 8.±2.6)·105 s-1, ΔH‡ = 46.(±4.6 ) kJ mol-1, ΔS‡ = 22(±18) J mol-1 K-1 and ΔV‡ = +1.85 (±0.1) cm3 mol-1 for waters bound to the fully protonated, neutral molecule. Regressing the pH variation of the experimental rate coefficient versus (1/[H+]) yields a similar value of kex298 = 8.3(±0.8)·105 s-1. These rates are ~104 times faster than those of the [Fe(OH2)6]3+ ion (kex298 =1.6·102 s-1 3) but are about an order of magnitude slower than other studied aminocarboxylate complexes, although these complexes have seven-coordinated Fe(III), not six as in the [Fe4(OH)2(hpdta)2(H2O)4]0(aq) molecule. As pH approaches pKa1, the rates decrease and a compensatory relation becomes evident between the experimental ΔH‡ and ΔS‡ values. Such variation cannot be caused by enthalpy from the deprotonation reaction and is not well understood. A correlation between <FeIII-OH2> bond lengths and the logarithm of kex298 is geochemically important because it could be used to estimate rate coefficients for geochemical materials for which only DFT calculations are possible. This molecule is the only neutral, oxo-bridged Fe(III) multimer for which rate data are available.
Panasci, A. F., Ohlin, C. A., Harley, S. J. and Casey, W. H. Rates of Water Exchange on the [Fe4(OH)2(hpdta)2(H2O)4]0 Molecule and Its Implicatiopns for Geochemistry. Inorg. Chem., 2012, 51, 6731-6738.
CSF Award
15th of March, 2012

Congratulations to Rene Johnson for receiving the Swiss Federal Institute of Technology - Centro Stefano Franscini - CSF award for best presentation by a young scientist at the "Uranium biogeochemistry: transformations and applications" international workshop in Ascona, Switzerland. She received this award for her presentation of her poster entitled "Reaction Kinetics of Aqueous Uranyl Complexes". The poster highlighted current projects as well as most reccently published work.

Olivia Allsman Joins the Lab!
10th of February, 2012

The Casey lab would like to welcome Olivia Allsman as the newest undergraduate member of the lab! Olivia is an undegraduate student at UC Davis majoring in both Geology and Chemistry.
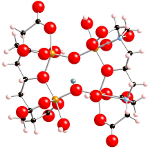
"Metastable structures and isotope
exchange reactions in polyoxometalate ions provide a molecular view of oxide dissolution" makes the news!
10th of January, 2012
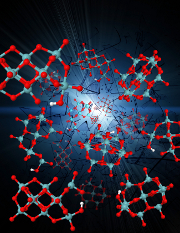
The article, published in Nature Materials (2012), is gathering attention in the online media.
The first online news article appeared on the UC Davis News and Information website then on ScienceDaily.com. A vdieo showing the interaction of between water and metal oxides will be available shortly.
Jungho Son joins the Casey lab!
December 2, 2012

The Casey lab's newest member! Jungho Son joins the lab as the newest research scientist.
Metastable structures and isotope
exchange reactions in polyoxometalate ions provide a molecular view of oxide dissolution
17th of November, 2011
Abstract: N/A
James R. Rustad and William H. Casey Metastable structures and isotope exchange reactions in polyoxometalate ions provide a molecular view of oxide dissolution. Nature Mater., 2012, 11, 223-226.
Katie Gronotte joins the Casey lab!
November 15, 2011

Katie Gronotte is the Casy lab's newest undergraduate reseracher.
Cooperation between bound waters
and hydroxyls in controlling isotope-exchange rates
24th of October, 2011
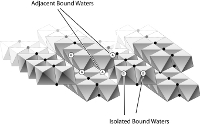
Abstract: Water molecules bound to mineral oxides differ from aqueous ions in that they are usually attached to different metal centers, or vicinal, and thus separated from one another. In contrast, for most monomeric ions used to establish kinetic reactivity trends, such as octahedral aquo ions [e.g., Al(H2O)63+], the bound waters are closely packed, or geminal. Because of this structural difference, the existing literature about ligand substitution in monomer ions may be a poor guide to the reactions of geochemical interest. To understand how coordination of the reactive functional groups might affect the rates of simple water-exchange reactions, we synthesized two structurally similar Rh(III) complexes, [Rh(phen)2(H2O)2]3+[1] and [Rh(phen)2(H2O)Cl]2+[2] where (phen)=1,10-phenanthroline. Complex [1] has two adjacent, geminal, bound waters in the inner-coordination sphere and [2] has a single bound water adjacent to a bound chloride ion. We employed Rh(III) as a trivalent metal rather than a more geochemically relevant metal like Fe(III) or Al(III) to slow the rate of reaction, which makes possible measurement of the rates of isotopic substitution by simple mass spectrometry. We prepared isotopically pure versions of the molecules, dissolved them into isotopically dissimilar water, and measured the rates of exchange from the extents of 18O and 16O exchange at the bound waters.
The pH dependency of rates differ enormously between the two complexes. Pseudo-first-order rates coefficients at 298 K for water exchanges from the fully protonated molecules are close: ![]() = 5·10-8(± 0.5·10-8) s-1 for [1] and
= 5·10-8(± 0.5·10-8) s-1 for [1] and ![]() = 2.5·10-9(± 1·10-9) for [2]. Enthalpy and entropy activation parameters (ΔH‡ and ΔS‡) were measured to be 119(± 3) kJ mol-1, and 14(±1) J mol-1 K-1, respectively for [1]. The corresponding parameters for the mono-aquo complex, [2], are 132(±3) kJ mol-1 and 41.5(±2) J mol-1 K-1. Rates increase by many orders of magnitude upon deprotonation of one of the bound waters in complex [1] because of the close proximity of a transferable proton that can convert the bound hydroxyl to a bound water. This interconversion allows the oxygen to exchange as a bound water, rather than as a bound hydroxyl, which is slow at near-neutral pH conditions.
= 2.5·10-9(± 1·10-9) for [2]. Enthalpy and entropy activation parameters (ΔH‡ and ΔS‡) were measured to be 119(± 3) kJ mol-1, and 14(±1) J mol-1 K-1, respectively for [1]. The corresponding parameters for the mono-aquo complex, [2], are 132(±3) kJ mol-1 and 41.5(±2) J mol-1 K-1. Rates increase by many orders of magnitude upon deprotonation of one of the bound waters in complex [1] because of the close proximity of a transferable proton that can convert the bound hydroxyl to a bound water. This interconversion allows the oxygen to exchange as a bound water, rather than as a bound hydroxyl, which is slow at near-neutral pH conditions.
Panasci, A. F., McAlpin, J. G., Ohlin, C. A., Christensen, S., Fettinger, J. C., Britt, R. D., Rustad, J. R., Casey, W. H. Cooperation between bound waters and hydroxyls in controlling isotope-exchange rates. Geochim. Cosmochim. Acta, 2012, 78, 18-27.
Hugues Jaccard joins the Casey lab!
September 30, 2011

The Casey lab has a new addition! Hugues Jaccard has joined us after completing his Ph.D. at the EPFL in Lausanne, Switzerland.
Multinuclear NMR study of the pressure
dependence for carbonate exchange in the UO2(CO3)34-(aq) ion
16th of September, 2011
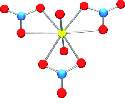
Abstract: N/A
Johnson, R. L., Harley, S. J., Ohlin, C. A., Panasci, A. F., Casey W. H. Multinuclear NMR study of the pressure
dependence for carbonate exchange in the UO2(CO3)34-(aq) ion. ChemPhysChem, 2011, 12, 2903-2906.
Electronic structure description of a
[Co(III)3Co(IV)O4 cluster: A model for the paramagnetic intermediate in cobalt-catalyzed water oxidation
13th of September, 2011
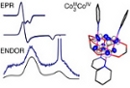
Abstract: Multifrequency electron paramagnetic resonace (EPR) spectroscopy and electronic structure calculations were performed on [Co4O4(C5H5N)4(CH3CO2)4]+ (1+), a cobalt tetramer with total electron spin S = 1/2 and formal cobalt oxidation states III, III, III, and IV. The cuboidal arrangement of its cobalt and oxygen atoms is similar to that of proposed structures for the molecular cobaltate clusters of the cobalt–phosphate (Co–Pi) water-oxidizing catalyst. The Davies electron-nuclear double resonance (ENDOR) spectrum is well-modeled using a single class of hyperfine-coupled 59Co nuclei with a modestly strong interaction (principal elements of the hyperfine tensor are equal to [-20(±2), 77(±1), -5(±15)] MHz). Mims 1H ENDOR spectra of 1+ with selectively deuterated pyridine ligands confirm that the amount of unpaired spin on the cobalt-bonding partner is significantly reduced from unity. Multifrequency 14N ESEEM spectra (acquired at 9.5 and 34.0 GHz) indicate that four nearly equivalent nitrogen nuclei are coupled to the electron spin. Cumulatively, our EPR spectroscopic findings indicate that the unpaired spin is delocalized almost equally across the eight core atoms, a finding corroborated by results from DFT calculations. Each octahedrally coordinated cobalt ion is forced into a low-spin electron configuration by the anionic oxo and carboxylato ligands, and a fractional electron hole is localized on each metal center in a Co 3dxz,yz-based molecular orbital for this essentially [Co+3.1254O4] system. Comparing the EPR spectrum of 1+ with that of the catalyst film allows us to draw conclusions about the electronic structure of this water-oxidation catalyst.
J. Gregory McAlpin, Troy A. Stich, C. André Ohlin, Yogesh Surendranath, Daniel G. Nocera, William H. Casey, R. David Britt Electronic structure description of a [Co(III)3Co(IV)O4 cluster: A model for the paramagnetic intermediate in cobalt-catalyzed water oxidation J. Am. Chem. Soc., 2011, 133, 15444-15452.
Electrochemical water oxidation with
cobalt-based electrocatalysts from pH 0-14: The thermodynamic basis for catalyst structure, stability and activity
1st of August, 2011

Abstract: Building upon recent study of cobalt-oxide electrocatalysts in fluoride-buffered electrolyte at pH 3.4, we have undertaken a mechanistic investigation of cobalt-catalyzed water oxidation in aqueous buffering electrolytes from pH 0-14. This work includes electrokinetic studies, cyclic voltammetric analysis, and electron paramagnetic resonance (EPR) spectroscopic studies. The results illuminate a set of interrelated mechanisms for electrochemical water oxidation in alkaline, neutral, and acidic media with electrodeposited Co-oxide catalyst films (CoOxcfs) as well as for a homogeneous Co-catalyzed electrochemical water oxidation reaction. Analysis of the pH dependence of quasi-reversible features in cyclic voltammograms of the CoOxcfs provides the basis for a Pourbaix diagram that closely resembles a Pourbaix diagram derived from thermodynamic free energies of formation for a family of Co-based layered materials. Below pH 3, a shift from heterogeneous catalysis producing O2 to homogeneous catalysis yielding H2O2 is observed. Collectively, the results reported here provide a foundation for understanding the structure, stability, and catalytic activity of aqueous cobalt electrocatalysts for water oxidation.
James B. Gerkent, J. Gregory McAlpin, Jaime Y. C. Chent, Matthew L. Rigsby, William H. Casey, R. David Britt, Shannon S. Stahl Electrochemical water oxidation with cobalt-based electrocatalysts from pH 0-14: The thermodynamic basis for catalyst structure, stability and activity J. Am. Chem. Soc., 2011, 133, 14431-14442.
"Water oxidation catalysis by manganese
in a geochemical-like cycle" makes the news
25th of May, 2011

The article, published in Nature Chemistry (2011, 3, 461-466), is gathering attention in the online media.
First, the UC Davis press release and blog. And here's what Chemistry News Articles, ScienceDaily.com, fuelcellsworks.com and R&D Mag have to say.
Geochemical kinetics
via the Swift-Connick equations and solution NMR
10th of April, 2011

Abstract: Signal analysis in Nuclear Magnetic Resonance spectroscopy is among the most powerful methods to quantify reaction rates in aqueous solutions. To this end, the Swift- Connick approximations to the Bloch-McConnell equations have been used extensively to estimate rate parameters for elementary reactions. The method is primarily used for 17O-NMR in aqueous solutions, but the list of geochemically relevant nuclei that can be used is long, and includes 29Si, 27Al, 19F, 13C and many others of particular interest to geochemists. Here we review the derivation of both the Swift-Connick and Bloch-McConnell equations and emphasize assumptions and quirks. For example, the equations were derived for CW-NMR, but are used with modern pulse FT-NMR and can be applied to systems that have exchange rates that are shorter than the lifetime of a typical pulse. The method requires a dilute solution where the minor reacting species contributes a neglible amount of total magnetization. We evaluate the sensitivity of results to this dilute-solution requirement and also highlight the need for chemically well- defined systems if reliable data are to be obtained. The limitations in using longitudinal relaxation to estimate reaction rate parameters are discussed. Finally, we provide examples of the application of the method, including ligand exchanges from aqua ions and hydrolysis complexes, that emphasize its flexibility. Once the basic requirements of the Swift-Connick method are met, it allows geochemists to establish rates of elementary reactions. Reactions at this scale lend themselves well to methods of computational simulation and could provide key tests of accuracy.
Harley, Stephen J.; Ohlin, C. André; Casey, William H. Geochemical kinetics via the Swift-Connick equations and solution NMR Geochim. Cosmochim. Acta , 2011, 75, 3711-3725
LinkWater oxidation catalysis by manganese
in a geochemical-like cycle
23rd of March, 2011
Abstract: Water oxidation in all oxygenic photosynthetic organisms is catalysed by the Mn4CaO4 cluster of Photosystem II. This cluster has inspired the development of synthetic manganese catalysts for solar energy production. A photoelectrochemical device, made by impregnating a synthetic tetranuclear-manganese cluster into a Nafion matrix, has been shown to achieve efficient water oxidation catalysis. We report here in situ X-ray absorption spectroscopy and transmission electron microscopy studies that demonstrate that this cluster dissociates into Mn(II) compounds in the Nafion, which are then reoxidized to form dispersed nanoparticles of a disordered Mn(III/IV)-oxide phase. Cycling between the photoreduced product and this mineral-like solid is responsible for the observed photochemical water-oxidation catalysis. The original manganese cluster serves only as a precursor to the catalytically active material. The behaviour of Mn in Nafion therefore parallels its broader biogeochemistry, which is also dominated by cycles of oxidation into solid Mn(III/IV) oxides followed by photoreduction to Mn2+.
Hocking, Rosalie K.; Brimblecombe, Robin; Chang, Shery L. Y.; Singh, Archana; Cheah, Mun Hoh; Glover, Chris; Casey, William H.; Spiccia, Leone Water oxidation catalysis by manganese in a geochemical-like cycle Nature Chem., 2011, 3, 461-466.
17O NMR and computational
study of a tetrasiliconiobate ion, [H2+xSi4Nb16O56](14-x)-.
23rd of March, 2011
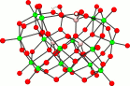
Abstract: Rates of oxygen-isotope exchange were measured in a tetrasiliconiobate ion in order to better understand how large oxide ions interact with water. The molecule has 19 non-equivalent oxygen sites and is sufficiently complex to evaluate hypotheses derived from our previous work on smaller clusters. We want to examine the extent to which individual oxygens react independently with particular attention given to the order of protonation of the various oxygen sites as the pH decreases from 13 to 6. As in our previous work, we find that the set of oxygen sites reacts at rates that vary over ca 104 across the molecule between pH 6 and 13 but with similar pH dependencies. There is NMR evidence of an intra- or intermolecular reaction at pH 7 where new peaks began to slowly form without losing the 17O isotopic tag and at pH less-or-equal-to 6 these new peaks formed rapidly. The oxygens bonded to silicon atoms began to isotopically exchange at pH 9 and below. The 17O NMR peak positions also vary considerably with pH for some, but not all, non-equivalent oxygen sites. This variation could be only partly accounted by electronic calculations, which indicate that oxygens should shift similarly upon protonation. Instead, we see that some sites change enormously with pH while other, similarly coordinated oxygens are less affected, suggesting that either some protons are exchanging so rapidly that the oxygen sites are seeing an averaged charge, or that counterions are modulating the ffect of the coordinated protons.
Johnson, Rene L.; Villa, Eric M.; Ohlin, C. André; Rustad, James R.; Casey, William H. 17O NMR and computational study of a tetrasiliconiobate ion, [H2+xSi4Nb16O56](14-x)- Chem. Eur. J., 2011 , 17, 9359-9367.
The pressure dependence of
oxygen-isotope-exchange rates between solution and apical oxygens on the UO2(OH)42- ion.
16th of February, 2011

Abstract: N/A
Harley, S. J., Ohlin, C. A., Johnson, R. L., Panasci, A., Casey W. H. The pressure dependence of oxygen-isotope-exchange rates between solution and apical oxygens on the UO2(OH)42- ion. Angew. Chem. Int. Ed. 2011, 50(19), 4467-4469.
Rates of water exchange for two cobalt(II) heteropolyoxotungstate
compounds in aqueous solution
13th of January, 2011

Abstract: Polyoxometalate ions are being used as ligands in water-oxidation processes related to solar energy production. An important step in these reactions must be the association and dissociation of water from the catalytic sites, the rates of which are unknown. Here we report the exchange rates of water ligated to Co(II) atoms in two polyoxotungstate sandwich molecules using the 17O-NMR-based Swift-Connick method. The compounds were the [Co4(H2O)2(B-a-PW9O34)2]10- and the larger abba−[Co4(H2O)2(P2W15O56)2]16- ions, each with two waters boundtrans to one another in a Co(II) sandwich between the tungstate ligands. The clusters, in both solid and solution state, were characterized by a range of methods, including NMR, ESI-MS, EXAFS, EPR, FT-IR, UV-Vis and potentiometry. For [Co4(H2O)2(B-a-PW9O34)2]10- at pH=5.4, we estimate: k298 = 1.55+/-0.3.106 s-1, ΔH = 39.8+/-0.4 kJ mol-1, ΔS = +7.1+/-1.2 J mol-1 K-1 and ΔV = 5.6 +/- 1.6 cm3.mol-1. For the Wells-Dawson sandwich cluster (abba−[Co4(H2O)2(P2W15O56)2]16-) at pH=5.54, we find: k298 = 1.62+/-0.3.106 s-1, ΔH =27.6+/-0.4 kJ mol-1 ΔS = -33+/-1.3 J mol-1 K-1 and ΔV = 2.2 +/- 1.4 cm3 .mol-1 at pH=5.2. The molecules are clearly stable and monospecific in slightly acidic solutions but dissociate in strongly acidic solutions. This dissociation is detectable via EPR spectroscopy as S=3/2 Co(II) species (such as as the [Co(H2O)6]2+ monomer ion) and by the significant reduction of the Co-Co vector in the XAS spectra.
C. A. Ohlin, S. J. Harley, J. G. McAlpin, R. K. Hocking, B. Q. Mercado, R. Johnson, E. M. Villa, M. K. Fidler, M. M. Olmstead, L. Spiccia, R. D. Britt, and W. H. Casey Rates of water exchange for two cobalt(II) heteropolyoxotungstate compounds in aqueous solution Chemistry - a European Journal , 2011, 17(16), 4408-4417
Adding reactivity to structure 2:
Oxygen-isotope-exchange rates in three isostructural oxide ions
December 6, 2010

Abstract: To understand how oxide structures react at the molecular scale, rates of steady oxygen-isotope exchanges were followed in three isostructural molecules of ca 40 atoms as a function of solution composition. These molecules were chosen because the structures in solution are known with complete confidence, yet isotope-exchange reactions can be followed spectroscopically at individual oxygens. The series of molecules differ only in a single Ti(IV) --> substitution in one of the three metal sites, making a series of structures having the stoichiometries: [HxNb
Villa, E. M., Ohlin C. A., Casey W. H. Adding reactivity to structure 2: Oxygen-isotope-exchange rates in three isostructural oxide ions American Journal of Science , 2010, 310, 629-644.
Isotopic Fractionation of Mg2+(aq),
Ca2+(aq), and Fe2+(aq) with Carbonate Minerals
August 25, 2010
Abstract Density functional electronic structure calculations are used to compute the equilibrium constant (the isotope fractionation factor) for 26Mg/24Mg and 44Ca/40Ca isotope exchange between carbonate minerals and uncomplexed divalent aquo ions. The most reliable calculations at the B3LYP/6-311++G(2d,2p) level predict equilibrium constants K, reported as 103ln(K) at 25 deg. C, of -5.3, -1.1, and +1.2 for 26Mg/24Mg exchange between calcite (CaCO3), magnesite (MgCO3), and dolomite (Ca0.5Mg0.5CO3), respectively, and Mg2+(aq), with positive values indicating enrichment of the heavy isotope in the mineral phase. For 44Ca/40Ca exchange between calcite and Ca2+(aq) at 25 deg. C, the calculations predict values of +1.5 for Ca2+(aq) in six-fold coordination and +4.1 for Ca2+(aq) in seven-fold coordination. We find that the reduced partition function ratios can be reliably computed from systems as small as M(CO3)610- and M(H2O)62+ embedded in a set of fixed atoms representing the 2nd shell (and greater) coordination environment. We find that the aqueous cluster representing the aquo ion is much more sensitive to improvements in the basis set than the calculations on the mineral systems, and that fractionation factors should be computed using the best possible basis set for the aquo complex, even if the reduced partition function ratio calculated with the same basis set is not available for the mineral system. The new calculations show that the previous discrepancies between theory and experiment for Fe3+-hematite and Fe2+-siderite fractionations arise from an insufficiently accurate reduced partition function ratio for the Fe3+(aq) and Fe2+(aq) species.
Rustad, J.R., Casey, W. H., Yin, Q.-Z., Bylaska, E. J., Felmy, A. R., Bogatko, S. A., Jackson, V. E., Dixon, D. A. Isotopic Fractionation of Mg2+(aq), Ca2+(aq), and Fe2+(aq) with Carbonate Minerals Geochim. Cosmochim. Acta, 2010, 47(22), 6301-6323
Adele Panasci joins the Casey lab
July 14, 2010

The Casey lab has a new addition in Adele Panasci who has joined us after completing her undergraduate degree at Saint Mary's College of California and a year working at the USDA in Alameda
Stumm medal 2010
June 27, 2010
Photos from the Goldschmidt conference at Oakridge:


The first peroxotitanoniobate cluster
- [N(CH3)4]10[Ti12Nb6O38(O2)6]
June 10, 2010
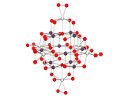
Abstract The first example of a discrete peroxo-polyoxotitanoniobate is introduced and characterized by x-ray crystallography.
Ohlin, C. A., Villa, E. M., Fettinger, J. C., Casey, William H. The first peroxotitanoniobate cluster - [N(CH3)4]10[Ti12Nb6O38(O2)6] Inorg. Chimica Acta, 2010, 363(15), 4405-4407.
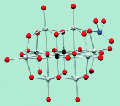
99Technetium-99 MAS NMR spectroscopy of a
cationic framework material that traps TcO4- ions
June 10, 2010
Abstract Not available.
Ping, Y., Wang, S., Alekseev, E.V., Depmeier, W., Hobbs, D.T., Albrecht-
Schmitt, T. E., Phillips, B. L., Casey, W. H. 99Tc-MAS-NMR on a Cationic Framework
Material for Trapping TcO4- Angew. Chem. Int. Ed., 2010, 49(34), 5975-5977.
Chris Colla joins the Casey Lab
June 2010

Chrisopher Colla from the geology department joins the Casey lab to participate in undergraduate research.
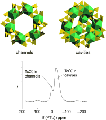
Borate Accelerates Oxygen-Isotope Exchange
for Polyoxoniobate Ions in Water
May 26, 2010
Abstract Understanding simple oxygen exchange reactions is important to a variety of communities concerned with the chemistry of oxides with water. Limitations in the methods available for studying reactions at these oxide-water interfaces, as well as difficulties in characterizing their structures, have led to the use of polyoxometalates (POMs) as model molecules. POMs are metaloxide ions comprised of group 5 and 6 metals. These ions constitute discrete and often soluble clusters than can be spectroscopically probed with great confidence. In addition, POMs are interesting in their own right owing to their structural and chemical diversity, and are finding an increasing number of applications. We have been investigating the oxygen-isotope exchange kinetics in these ions and aqueous solution by 17O Nuclear Magnetic Resonance (NMR) to help better recognize what controls molecule-water interface processes on the level of individual oxygen sites. These structures are chosen because the isotope-exchange reactions could be followed separately from dissociation or condensation of the structure.
Villa, E. M., Ohlin, C. A, Casey, W. H..Borate Accelerates Oxygen-Isotope Exchange for Polyoxoniobate Ions in Water Eur. Chem. J. , 2010, 16(29), 8631-8634.
How the overlapping time scales for
peptide binding and terrace exposure lead to nonlinear step dynamics during growth of calcium oxalate monohydrate
May 21, 2010

Abstract Using in-situ atomic force microscopy (AFM), we investigate the inhibition of calcium oxalate monohydrate (COM) step growth by aspartic acid-rich peptides and find that the magnitude of the effect depends on terrace lifetime. We then derive a time-dependent step-pinning model in which average impurity spacing depends on the terrace lifetime as given by the ratio of step spacing to step speed. We show that the measured variation in step speed is well fit by the model and allows us to extract the characteristic peptide adsorption time. The model also predicts that a crossover in the time scales for impurity adsorption and terrace exposure leads to bistable growth dynamics described mathematically by a catastrophe. We observe this behaviour experimentally both through the sudden drop in step speed to zero upon decrease of supersaturation and through fluctutations in step speed between the two limiting values at the point where the catastrophe occurs. We discuss the model's general applicability to macromolecular modifiers and biomineral phases.
Weaver, M. L., Qiu, S. R., Friddle, R. W., Casey, W. H., De Yoreo, J. J. How the overlapping time scales for peptide binding and terrace exposure lead to nonlinear step dynamics during growth of calcium oxalate monohydrate Crystal growth & design , 2010, 10(7), 2954–2959. Link
C&EN: Cobalt(IV) caught in water-splitting action
May 7, 2010
The recent article in JACS by Greg McAlpin as part of a three-way collaboration between W. H. Casey (UCD), R. D. Britt (UCD) and D. Nocera (MIT), is featured in C&EN.
Drahl, C. Cobalt(IV) caught in water-splitting action C&EN , 2010,88(19), 36-37.
EPR evidence for Co(IV) species produced
during water oxidation at neutral pH
April 21, 2010
Abstract Thin film water oxidation catalysts (Co-Pi) prepared by electrodeposition from phosphate electrolyte and Co(II) nitrate have been characterized by electron paramagnetic resonance (EPR) spectroscopy. Co-Pi catalyst films exhibit EPR signals corresponding to populations of both Co(II) and Co(IV). As the deposition voltage is increased into the region where water oxidation prevails, the population of Co(IV) rises and the population of Co(II) decreases. The changes in the redox speciation of the film can also be induced, in part, by prolonged water oxidation catalysis in the absence of additional catalyst deposition. These results provide spectroscopic evidence for the formation of Co(IV) species during water oxidation catalysis at neutral pH.
McAlpin, J. G., Surendranath, Y., Dinca, M., Stich, T., Stoian, S., Casey, W. H., Nocera, D., Britt, R. D. EPR Evidence for Co(IV) Species Produced During Water Oxidation at Neutral pH J. Am. Chem. Soc., 2010,132(20), 6882-6883.
NSF fellowship
April 6, 2010

Congratulations to Rene Johnson for being awarded an NSF Graduate Research Fellowship yesterday!
Graduation
April 2, 2010

Congratulations to Eric Villa for finishing his PhD this week! He will remain as a postdoctoral researcher until June when he moves to take up a new position in the group of Thomas Albrecht-Schmidt at the University of Notre Dame.
Oxygen-isotope exchange rates for three
isostructural polyoxometalate ions
March 8, 2010

Abstract We compare oxygen-isotope exchange rates for all structural oxygens in three polyoxoniobate ions that differ by systematic metal substitutions of Ti(IV)=>Nb(V). The [HxNb10O28 ](6-x)-, [HxTiNb9O28](7-x)-, and [HxTi2 Nb8O28](8-x)- ions are all isostructural, yet have different Brönsted properties. Rates for sites within a particular molecule in the series differ by at least 104, but the relative reactivities of the oxygen sites rank in nearly the same relative order for all ions in the series. Within a single ion, most structural oxygens exhibit rates of isotopic exchange that vary similarly with pH, indicating that each structure responds as a whole to changes in pH. Across the series of molecules, however, the pH dependencies for isotope exchanges and dissociation are distinctly different, reflecting different contributions from proton- or base-enhanced pathways. The proton-enhanced pathway for isotope exchange dominates at most pH conditions for the [HxTi2 Nb8O28](8-x)- ion, but the base-enhanced pathways are increasingly important for the [HxTiNb9O28](7-x)- and [HxNb10O28](6-x)- structures at higher pH. The local effect of Ti(IV) substitution could be assessed by comparing rates for structurally similar oxygens on each side of the [HxTiNb9O28](7-x)- ion and is surprisingly small. Interestingly, these nanometer-size structures seem to manifest the same general averaged casey +geo amphoteric chemistry that is familiar for other reactions affecting oxides in water, including interface dissolution by proton- and hydroxyl-enhanced pathways.
Villa, E. M., Ohlin, C. A., Casey, W. H. Oxygen-isotope exchange rates for three isostructural polyoxometalate ions J. Am. Chem. Soc., 2010,132(4), 5264-5272.
William H. Casey is awarded the 2010 Science
Innovation Award by the EAG
March 7, 2010

The 2010 EAG Science Innovation Award has been named in honour of Werner Stumm for advances in low temperature geochemistry and/or processes at the mineral-fluid interface. The recipient of the 2010 Science Innovation Award is William H. Casey.
NDTB-1: A supertetrahedral cationic framework
that removes TcO4- from solution
March 7, 2010

Abstract A cubic thorium borate possesses a porous supertetrahedral cationic framework with extraframework borate anions. These anions are readily exchanged with a variety of environmental contaminants (see picture), especially those from the nuclear industry, including chromate and pertechnetate.
Wang, S.; Alekseev, V.; Diwu, J.; Casey, W.H.; Phillips, B.L.; Depmeier, W.; Albrecht-Schmitt, T.E "NDTB-1: A supertetrahedral cationic framework that removes TcO4- from solution ", Angew. Chem. Int. Ed, 2010, 49(6) , 1057-1060.
Porous Capsules {(M)M5}12FeIII30
(M = MoVI, WVI):
Sphere Surface
Supramolecular Chemistry with 20 Ammonium Ions, Related Solution
Properties, and Tuning of Magnetic Exchange Interactions
March 7, 2010

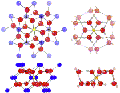
Abstract A manifold of hydrogen-bonding options on a highly active, functionalized capsule surface exhibiting 20 crown ether type pores allows the fixation and recognition of 20 ammonium cations (see picture; W green, Fe orange, O red, N blue, H light gray), which are partially released in solution, thereby leading to related equilibria.
Todea, A.M.; Merca, A.; Bögge, H.; Glaser, T.;
Pigga, J.M.; Langston, M.L.K.; Liu, T; Prozorov, R.; Luban, M;
Schröder, C.; Casey, W.H.; Müller, A.![]() "Porous Capsules {(M)M5}12FeIII30
(M = MoVI, WVI): Sphere Surface
Supramolecular Chemistry with 20 Ammonium Ions, Related Solution
Properties, and Tuning of Magnetic Exchange Interactions", Angew.
Chem. Int. Ed., 2010, 49(3), 514-519.
"Porous Capsules {(M)M5}12FeIII30
(M = MoVI, WVI): Sphere Surface
Supramolecular Chemistry with 20 Ammonium Ions, Related Solution
Properties, and Tuning of Magnetic Exchange Interactions", Angew.
Chem. Int. Ed., 2010, 49(3), 514-519.